
外傷性脳損傷(TBI)は、事故などによる頭部への物理的な衝撃によって引き起こされ、しばしば運動機能に永続的な障害を残します。特に慢性期における運動麻痺は、患者のQOL(生活の質)を著しく低下させるにもかかわらず、これまで有効な治療法が限られており、大きなアンメット・メディカル・ニーズ(未だ満たされていない医療ニーズ)が存在していました。この課題に対し、サンバイオ株式会社が開発した「アクーゴ®(一般名:バンデフィテムセル)」が、TBIに伴う慢性期運動麻痺の改善を目的とする日本初の再生医療等製品として承認されました。本稿では、この画期的な治療法のPMDA(独立行政法人 医薬品医療機器総合機構)承認情報の要点を、専門家向けに分かりやすく解説します。
P1: アクーゴ®(バンデフィテムセル)PMDA承認情報サマリー
アクーゴ®は、外傷性脳損傷(TBI)後の慢性期に固定化した運動麻痺の改善を目指す、国内で初めて承認された再生医療等製品です。この承認は、リハビリテーション以外に有効な治療選択肢がなかった患者とその家族にとって、機能回復への新たな希望をもたらす重要な一歩を意味します。
アクーゴ®(バンデフィテムセル) PMDA承認情報サマリー
• 製造販売元:サンバイオ株式会社
この革新的な治療法の登場は、TBI治療におけるパラダイムシフトを引き起こす可能性を秘めています。次章からは、その承認を支える科学的根拠と製品特性について、キーポイントを一つずつ詳述していきます。
P2: アクーゴ®承認のキーポイント

アクーゴ®の承認内容を理解するためには、その科学的背景から臨床的価値、そして市場導入の枠組みまでを網羅的に把握することが不可欠です。ここでは、その全体像を捉える上で戦略的に重要な「製品概要」「効能・効果」「主要な臨床成績」「承認ステータス」という4つの柱に焦点を当てて解説します。
• 1. 製品概要 (Product Profile): アクーゴ®は、健康な成人から採取した骨髄液を原料とする、ヒト(同種)骨髄由来の間葉系幹細胞を用いた再生医療等製品です。その技術的特徴として、Notch-1タンパク質の細胞内ドメインをコードする遺伝子が導入されており、定位脳手術により脳内の損傷部位周辺に直接移植して使用されます。
• 2. 効能・効果 (Indication): 本製品の適応は「外傷性脳損傷(TBI)による慢性期の運動麻痺の改善」です。これまで回復が困難とされてきた慢性期の機能障害に直接アプローチする、新しい治療コンセプトに基づいています。
• 3. 主要な臨床成績 (Key Clinical Results): 承認の科学的根拠となったのは、国際共同で実施された第Ⅱ相臨床試験です。この試験において、主要評価項目である運動機能評価尺度(FMMSスコア)で、偽手術群と比較して統計学的に有意な改善を達成しました。
• 4. 承認ステータス (Approval Status): 本製品は「7年間の条件及び期限付承認」という枠組みで承認されました。これは、臨床試験の成績が限られていることを踏まえ、画期的な医薬品を早期に患者へ届けるための制度です。そのため、市販後には使用した全症例を対象とした調査が義務付けられ、実臨床下での有効性と安全性をさらに検証していくことになります。
これらのキーポイントは、アクーゴ®がどのような製品であり、いかにして臨床応用への道を切り拓いたかを明確に示しています。次に、この治療法を構成する具体的なシステムについて掘り下げます。
P3: 細胞剤、専用投与機器、調製液から成る包括的な治療システム

アクーゴ®は、単なる細胞製剤にとどまりません。それは、治療効果の源泉である「細胞剤」、精密な投与を可能にする「専用投与機器」、そして移植準備に不可欠な「専用調製液」という3つの要素が一体となった包括的な「治療システム」です。この統合されたアプローチは、複雑な脳内移植手技の標準化と安全性の確保に大きく貢献します。
• 脳内移植用細胞剤 (Cell Product):
◦ 本質: ヒト(同種)骨髄由来間葉系幹細胞であり、開発コード「SB623」として知られています。
◦ 特徴: Notch-1タンパク質の細胞内ドメインをコードする遺伝子が導入されています。
◦ 提供形態: 1回の治療あたり4バイアル(各12.5×10⁶個の生細胞)が提供されます。
• 専用投与機器セット (Dedicated Device Set):
◦ 目的: 定位脳手術装置と組み合わせて使用し、細胞を脳内の標的部位へ正確に移植するために設計されています。
◦ 構成: マイクロシリンジ、投与カニューラ、インサーター、スタイレット、ガイド&ストップから構成されます。
• 専用調製液 (Preparation Solution):
◦ 用途: 凍結保存された細胞剤を融解後、移植に適した状態にするための洗浄、懸濁、調製に使用されます。
このシステム全体が連携することで、高度な再生医療の安全かつ効果的な実施が支援されます。次章では、特に精密な投与を実現するデバイスに焦点を当てて解説します。
P4: 高精度な脳内移植を実現する専用設計のデバイス
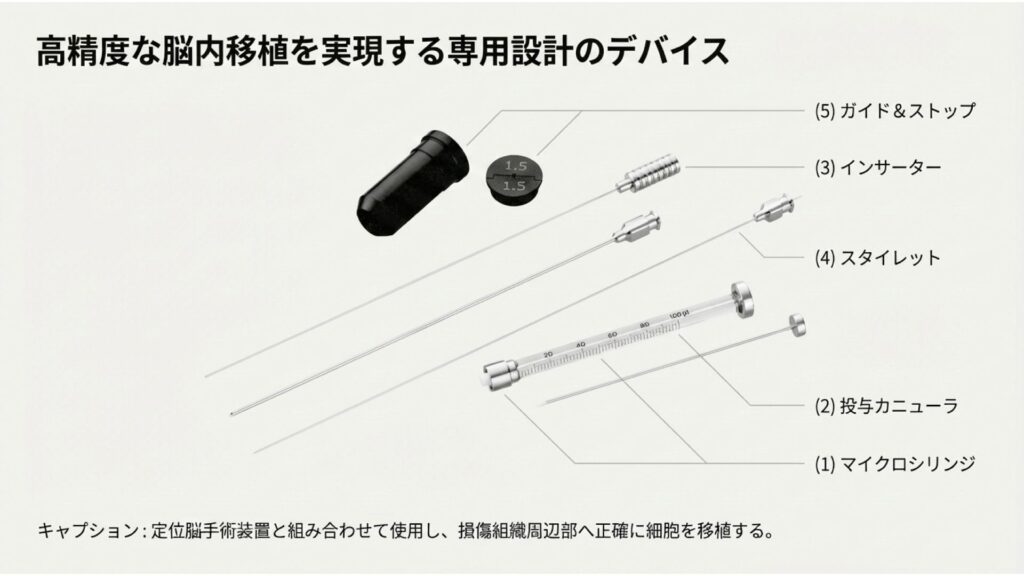
脳という極めて繊細な組織への細胞移植では、治療効果を最大化し、周辺組織へのダメージを最小化するために、極めて高い精度での投与が求められます。アクーゴ®の専用投与機器セットは、この厳しい要求に応えるために専門的に設計された医療機器です。
キャプション: 定位脳手術装置と組み合わせて使用し、損傷組織周辺部へ正確に細胞を移植する。
このデバイスは、以下の5つの主要なコンポーネントで構成されています。
1. マイクロシリンジ: 細胞懸濁液の精密な微量吸引と、制御された注入を行うためのシリンジ。
2. 投与カニューラ: 細胞を脳の標的深度まで導くための細径チューブ。
3. インサーター: カニューラを脳内に正確に挿入するための補助器具。
4. スタイレット: カニューラ挿入時に組織の削り取り(ティッシュ・コアリング)を防ぎ、内腔の閉塞を防止するために挿入される固形の芯。
5. ガイド&ストップ: カニューラを予め計画された標的深度まで正確に到達させ、それ以上進むことを防ぐための調節機構。
これらの精密なコンポーネントが一体となって機能することで、計画された標的部位への極めて正確な細胞移植が実現されます。それでは、このデバイスを用いて治療を受ける対象患者と具体的な投与方法はどのようになっているのでしょうか。
P5: 対象患者と用法・用量

あらゆる治療法の成功は、適切な患者を選択し、定められた用法・用量を遵守することにかかっています。特にアクーゴ®のような先進的な再生医療においては、その有効性と安全性を担保するため、これらの基準が厳密に定められています。
対象患者 (Target Patients)
アクーゴ®の投与対象となる患者は、以下の基準を満たす必要があります。
• 基本条件: 外傷性脳損傷の受傷後6カ月以上が経過し、リハビリテーション等を行っても運動機能障害が改善しない「固定期」にある患者。
◦ 注記:承認の根拠となった第Ⅱ相臨床試験(TBI-01)では、受傷後12カ月以上が経過した患者が組入れ対象でした。この点は、エビデンスを解釈する上で重要です。
• 重症度: GOS-E(Glasgow Outcome Scale Extended)スコアが3~6の中等度又は重度の患者。
• 画像診断: MRI等の画像診断で、現在の運動麻痺の原因となっている局所的な脳損傷が確認できる患者。
• 除外基準: 脳腫瘍のある患者、またはその既往歴のある患者については、本品の使用の可否を慎重に判断する必要があります。
用法・用量 (Administration & Dosage)
具体的な投与方法は、以下の通りです。
• 投与細胞数: 1回の治療で、生細胞として5×10⁶個(300μLの細胞懸濁液に調製)。
• 投与方法: 専用投与機器セットを用いた定位脳手術により、約10μL/minの注入速度で実施されます。
• 投与部位: 損傷した組織そのものではなく、その周辺部(皮質下の外傷近傍組織)に移植します。移植部位は、脳血管系、脳溝及び脳室を避けて慎重に選択されます。
• 投与経路: 頭蓋骨に開けた1箇所の小さな孔から、3つの異なる移植経路を設定。1つの経路あたり100μLの細胞懸濁液を、5箇所にそれぞれ20μLずつ、合計15箇所に分割して注入します。
これらの厳格な基準は、治療の再現性と安全性を確保するための基盤となります。次に、これらの基準に基づいて行われた有効性検証試験の結果を詳しく見ていきましょう。
P6: 有効性の検証:国際共同第Ⅱ相臨床試験(TBI-01)
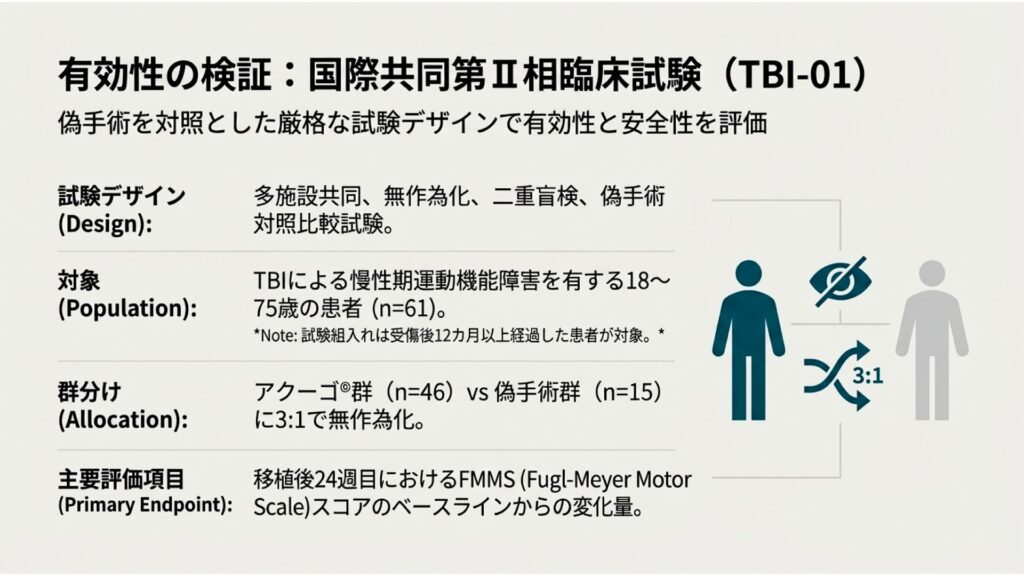
医薬品の有効性と安全性を科学的に証明するためには、質の高い臨床試験デザインが不可欠です。アクーゴ®の有効性を評価したTBI-01試験は、**「偽手術(シャム手術)を対照とした厳格な試験デザイン」**を採用しました。これは、被験者が実際の治療を受けたか否かを知ることができないため、プラセボ効果を排除し、治療法そのものの真の効果を客観的に評価できるゴールドスタンダードな手法です。この厳格な試験から得られたエビデンスが、承認における信頼性の高い根拠となりました。
• 試験デザイン (Design): 多施設共同、無作為化、二重盲検、偽手術対照比較試験。
• 対象 (Population): 外傷性脳損傷による慢性期の運動機能障害を有する18~75歳の患者61名。 注:試験組入れは、受傷後12カ月以上が経過した患者が対象でした。
• 群分け (Allocation): 患者は、アクーゴ®を投与する群(n=46)と、頭皮の切開のみを行う偽手術群(n=15)に、3:1の比率で無作為に割り付けられました。
• 主要評価項目 (Primary Endpoint): 治療介入(移植または偽手術)から24週目における、運動機能評価尺度FMMS(Fugl-Meyer Motor Scale)スコアの、治療開始前(ベースライン)からの変化量。
このような厳格なデザインの試験を通じて、アクーゴ®の有効性が検証されました。次のセクションでは、その最も重要な結果である主要評価項目の達成について具体的に解説します。
P7: 主要評価項目:アクーゴ®投与群で運動機能の有意な改善を達成
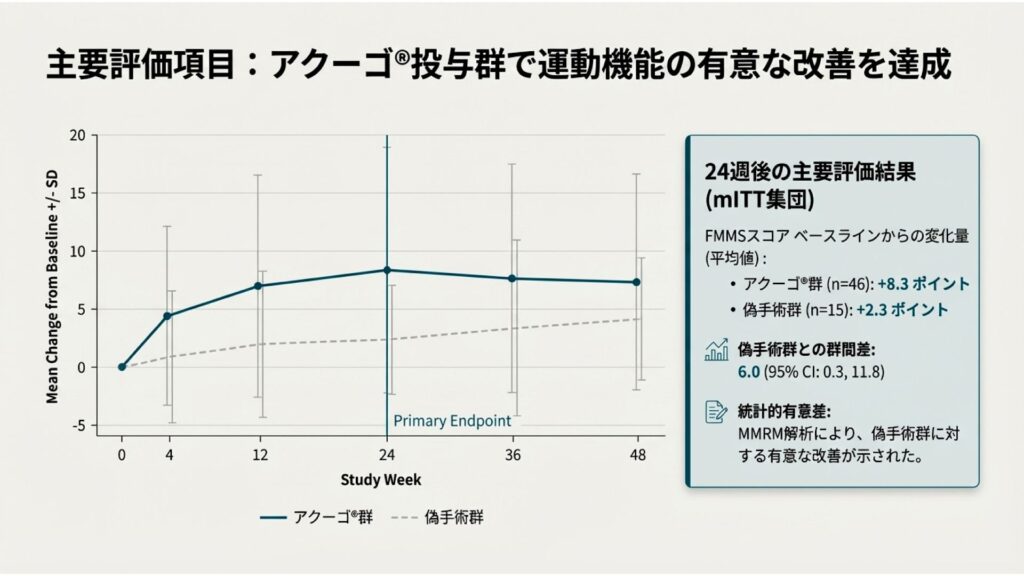
臨床試験の成否を判断する上で最も重要な指標は、事前に設定された主要評価項目の結果です。アクーゴ®は、この主要評価項目を見事に達成し、TBIによる慢性期運動麻痺に対する改善効果を客観的に証明しました。これが、PMDA承認の決定的な要因となりました。
24週後における主要評価結果(mITT集団)は以下の通りです。
• FMMSスコアのベースラインからの変化量(平均値):
◦ アクーゴ®群 (n=46): +8.3 ポイント
◦ 偽手術群 (n=15): +2.3 ポイント
• 偽手術群との群間差: 6.0 ポイント (95% CI: 0.3, 11.8)
• 統計的有意差: MMRM(Mixed Model for Repeated Measures)解析により、アクーゴ®群は偽手術群に対して統計学的に有意な改善を示しました(p=0.0037)。
グラフが示すように、運動機能の改善効果は主要評価項目である24週時点だけでなく、その後の48週まで持続する傾向が見られました。この統計学的に有意かつ臨床的にも意義のある改善結果は、アクーゴ®の有効性を裏付ける強力なエビデンスです。有効性の次は、安全性プロファイルについて検証します。
P8: 安全性プロファイル:主な副作用
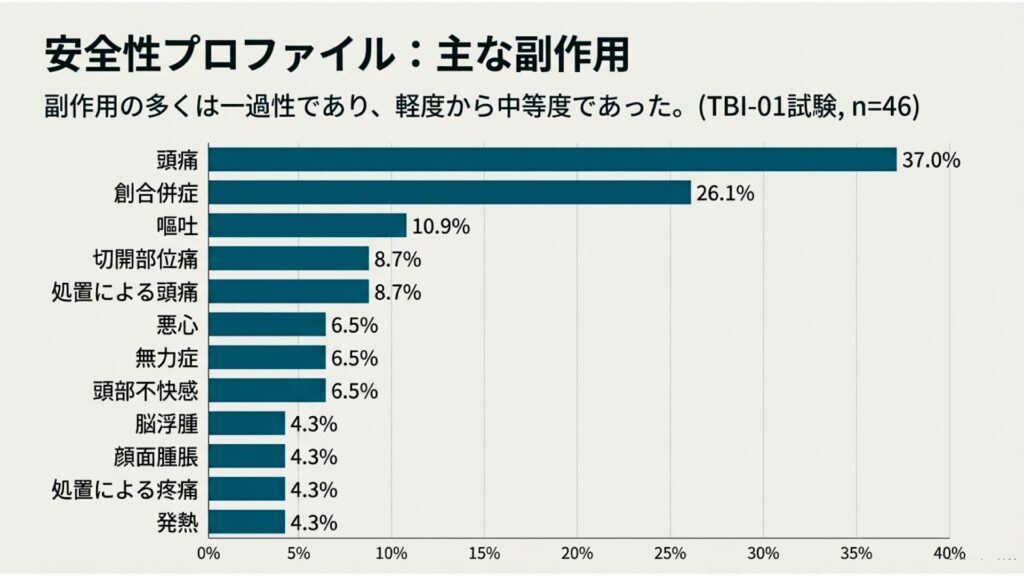
新しい治療法の評価において、有効性と並んで安全性の評価は極めて重要です。TBI-01試験(アクーゴ®群, n=46)で認められた副作用の多くは一過性であり、重症度は軽度から中等度でした。これは、アクーゴ®が全体として許容可能な安全性プロファイルを持つことを示唆しています。
報告された主な副作用は以下の通りです。
• 頭痛 (37.0%)
• 創合併症 (26.1%)
• 嘔吐 (10.9%)
• 切開部位痛 (8.7%)
• 処置による頭痛 (8.7%)
• 悪心 (6.5%)
• 無力症 (6.5%)
• 頭部不快感 (6.5%)
• 脳浮腫 (4.3%)
• 顔面腫脹 (4.3%)
• 処置による疼痛 (4.3%)
• 発熱 (4.3%)
これらの副作用の多くは、定位脳手術という頭蓋内への侵襲的な手技に起因するものと考えられ、全体として管理可能であることが示唆されます。しかし、より注意を要する重大な副作用と使用上の注意点についても理解しておく必要があります。
P9: 重大な副作用と使用上の重要な注意

治療法の安全性を包括的に理解するためには、頻度の高い副作用だけでなく、発生頻度は低いものの重篤化する可能性のある事象についても正確に把握することが不可欠です。ここでは、アクーゴ®の使用に伴う重大な副作用と、リスクを管理するための重要な注意事項について解説します。
重大な副作用 (Serious Adverse Events)
以下の重大な副作用が報告されています。
• 痙攣発作 (Seizure): 2.2%
• 出血 (Hemorrhage, 頭蓋内出血): 4.3%
• 譫妄 (Delirium): 2.2%
• 平衡障害 (Balance Disorder): 2.2%
• 感染症 (Infection): 頻度不明
重要な基本的注意 (Important Precautions)
安全な使用を徹底するため、以下の点に留意する必要があります。
• 生物由来原料のリスク: 本品は健康な成人ドナーの骨髄液を原料としています。ドナーは適格性確認のための問診や、HIV、HBV、HCV、HTLV等の感染症検査をクリアしていますが、現在の科学技術では検知できないウイルス等による感染症伝播のリスクを完全には排除できません。
• 患者への説明と同意: 本品が「条件及び期限付承認」であること、定位脳手術に伴うリスクを含め、十分な情報を文書で患者に説明し、文書による同意を得ることが必須です。
• 出血リスクの観察: 頭蓋内出血があらわれる可能性があるため、術後は適宜、頭部MRI又はCTを実施し、患者の状態を十分に観察する必要があります。
これらのリスク情報を医療従事者と患者が共有し、適切な対策を講じることが、アクーゴ®の安全な導入の鍵となります。次に、この製品が受けた特殊な承認制度について解説します。
P10: 「条件及び期限付承認」の理解

再生医療のような画期的な技術をいち早く患者に届けるため、日本の医薬品承認制度には「条件及び期限付承認」という特別な枠組みが存在します。この制度は、有効性が期待され、かつ緊急性の高い医薬品について、早期の実用化を可能にする一方で、市販後に厳格なデータ収集を義務付けることで、安全性と有効性を継続的に確認するものです。
アクーゴ®はこの制度の下で承認され、以下の期限と条件が課されています。
• 承認期限 (Time Limit): 7年間
• 主な承認条件 (Key Conditions):
1. 施設・医師要件: 外傷性脳損傷(TBI)の治療と定位脳手術に十分な知識・経験を持つ医師が、緊急時に迅速に対応できる医療施設でのみ使用すること。
2. 製造販売後調査: 本品を使用する全症例を対象とした調査を実施し、市販環境下での有効性・安全性に関するデータを収集・評価すること。
3. 品質・作用機序: 作用機序に関する情報を継続的に収集し、それに基づいて品質管理戦略を改良すること。
この承認制度は、イノベーションを促進しつつ、患者の安全を最優先に確保するための責任あるアプローチと言えます。次に、現時点で考えられているアクーゴ®の作用機序について考察します。
P11: 作用機序と体内動態に関する考察
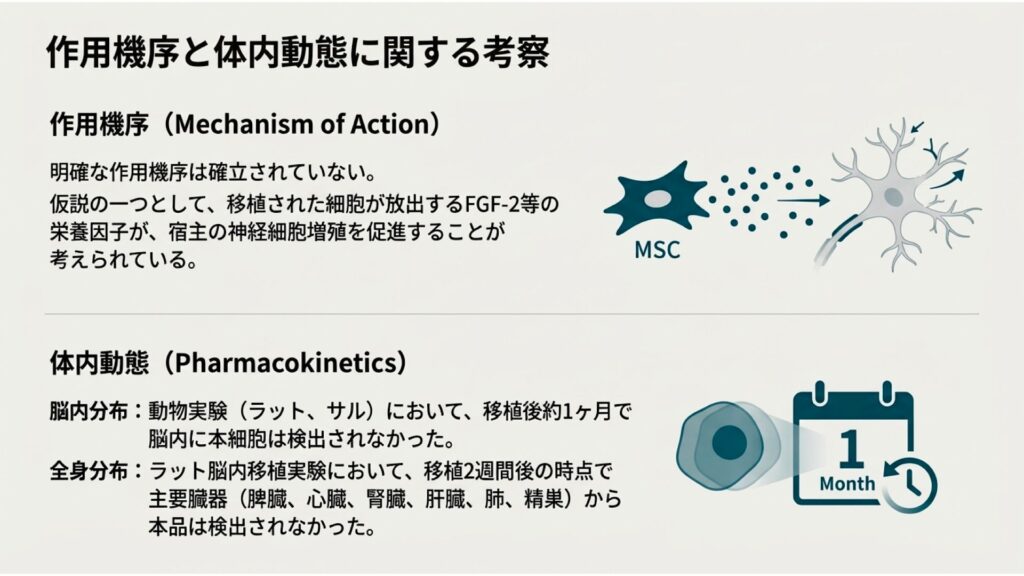
治療薬が「どのように効くか(作用機序)」そして「体内でどうなるか(体内動態)」を理解することは、その適正使用と将来的な発展のために不可欠です。ここでは、アクーゴ®に関して現時点で判明している科学的考察を解説します。
作用機序 (Mechanism of Action)
現時点で、アクーゴ®の明確な作用機序は確立されていません。しかし、研究から以下のような仮説が立てられています。
• 仮説: 脳内に移植された細胞が、細胞傷害や代謝ストレスなどにより**死滅する過程で、FGF-2(線維芽細胞増殖因子-2)**などの神経栄養因子を放出します。これらの因子が、宿主(患者自身)の神経細胞の増殖や再生を促進し、失われた神経ネットワークの再構築を助けることで、運動機能の改善に繋がるのではないかと考えられています。
体内動態 (Pharmacokinetics)
動物実験からは、移植された細胞の体内での動態について以下の知見が得られています。
• 脳内分布: ラットおよびサルの脳内に本品を移植した実験では、移植後約1ヶ月で脳内に本細胞は検出されなくなりました。
• 全身分布: ラットの脳内移植実験において、移植2週間後の時点で脾臓、心臓、腎臓、肝臓、肺、精巣といった主要な臓器から本品は検出されませんでした。
これらの知見は、移植された細胞が永続的に生着して機能するのではなく、一定期間、神経再生を促すシグナルを放出した後、体内から消失する「ヒット・アンド・ラン」のようなメカニズムで作用する可能性を示唆しています。
P12: アクーゴ®:TBI慢性期運動麻痺に対する新たな治療選択肢

これまでの情報を統合すると、アクーゴ®がTBI治療における新たな選択肢として持つ3つの重要な価値が浮かび上がります。それは「革新性」「エビデンス」「責任ある導入」です。この製品は単なる新薬ではなく、これまで治療を諦めざるを得なかった患者に対し、機能回復という新たな目標を提示する、治療戦略そのものに影響を与える可能性を秘めています。
• 革新性 (Innovation): 外傷性脳損傷後の運動機能障害を改善する、本邦で初めて承認された再生医療等製品であり、慢性期の神経障害に対する治療の扉を開きました。
• エビデンス (Evidence): 偽手術を対照とした厳格な第Ⅱ相臨床試験において、統計学的に有意な運動機能の改善効果と、許容可能な安全性プロファイルを示し、その臨床的価値を科学的に証明しました。
• 責任ある導入 (Responsible Introduction): 7年間の条件及び期限付承認の下、全例調査を通じてさらなるエビデンスを構築し、適正使用を推進するという、イノベーションと安全性の両立を目指す慎重かつ責任あるプロセスで導入されます。
この3つの柱が、アクーゴ®の臨床的価値と将来性を示しており、TBIに苦しむ多くの患者にとって、新たな希望の光となることが期待されます。
——————————————————————————–
注記
本資料は、再生医療等製品「アクーゴ®脳内移植用細胞剤」の独立行政法人医薬品医療機器総合機構(PMDA)公開情報に基づき作成されています。

コメント